
1-click AWS Deployment 1-click Azure Deployment
Overview
Avogadro has advanced an unconventional molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It offers flexible, high quality rendering, and a powerful plugin architecture. Characteristic uses include building molecular structures, formatting input files, and analyzing output of an extensive variety of computational chemistry packages. By using the CML file format as its native document type, Avogadro pursues to increase the semantic accessibility of chemical data types.
The work presented here details the Avogadro library, which is a framework providing a code library and application programming interface (API) with three-dimensional visualization capabilities; and has direct applications to research and education in the fields of chemistry, physics, materials science, and biology. The Avogadro application provides a rich graphical interface using dynamically loaded plugins through the library itself. The application and library can each be extended by implementing a plugin module in C++ or Python to explore different visualization techniques, build/manipulate molecular structures, and interact with other programs. We describe some example extensions, one which uses a genetic algorithm to find stable crystal structures, and one which interfaces with the PackMol program to create packed, solvated structures for molecular dynamics simulations. The 1.0 release series of Avogadro is the main focus of the results discussed here.
Avogadro offers a semantic chemical builder and platform for visualization and analysis. For users, it offers an easy-to-use builder, integrated support for downloading from common databases such as PubChem and the Protein Data Bank, extracting chemical data from a wide variety of formats, including computational chemistry output, and native, semantic support for the CML file format. For developers, it can be easily extended via a powerful plugin mechanism to support new features in organic chemistry, inorganic complexes, drug design, materials, biomolecules, and simulations. Avogadro is freely available under an open-source license from http://avogadro.openmolecules.net.
Avogadro 2
Avogadro 2 is a chemical editor and visualization application, it is also a set of reusable software libraries written in C++ using principles of modularity for maximum reuse. The development of the first generation Avogadro application and library is documented in our paper, and this remains the preferred method of citation at present. The motivation for rewriting Avogadro, along with improvements and changes made in Avogadro 2 are summarized in our Source article. We provide a set of permissively licensed, open source, cross platform software components in the Avogadro 2 libraries, along with an end-user application with full source code, and binaries.
The library features updated and improved rendering, where we built upon the abstraction provided by previous API, but implemented a simple scene graph. This makes use of features such as impostor sphere rendering, resulting in significant rendering speed improvements while improving the quality of the visualization. The core is built for scalability, looking to enable the analysis of larger chemical structures and simulations being produced by computational chemistry codes today. Emphasis has also been placed on making it even easier to extend, using simple Python scripts to add simulation input capabilities, and data input/output along with access to full-blown C++ plugin APIs where more control is required.
Avogadro is now able to make full use of the visualization capabilities of VTK, in addition to its own powerful rendering capabilities. This means that complex visualization, involving techniques such as volume rendering for point data, or streamlines for vector fields, will now become possible. The project is composed of two separate repositories, with the `avogadroapp’ repository offering a full demonstration of how to use the libraries in an end-user application. The `avogadrolibs’ repository contains all of the libraries, with the option to only build subsets. The development process uses distributed version control (Git), testing (CTest/CDash), and automated binary generation.
Molecule Modeling with Avogadro
.First, click on the “Element” button on the left and choose the “other” option to open up a periodic table of elements. From there you can click any element and then insert it on the drawing space by simply clicking at any point.
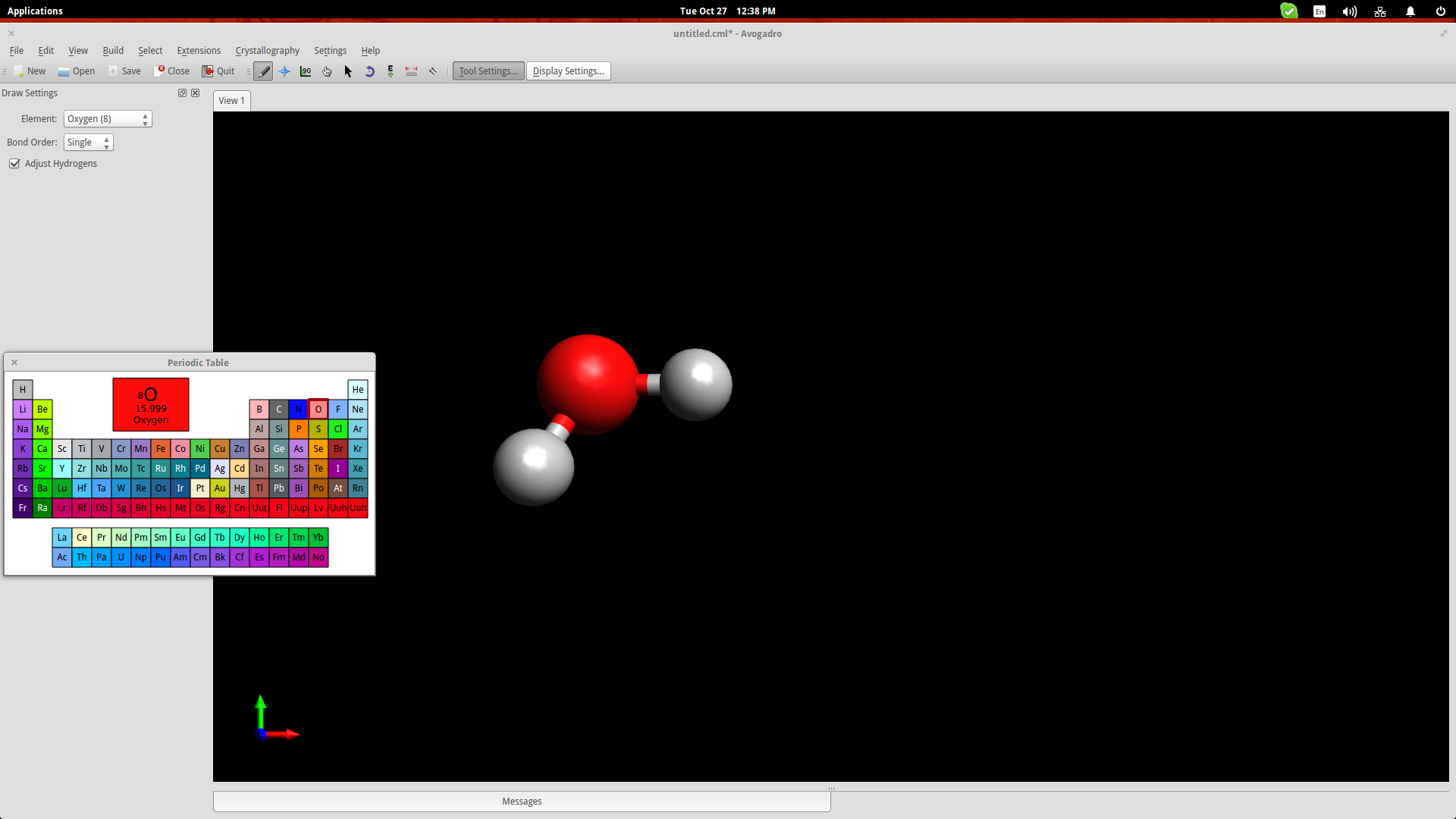
Avogadro has automatically bonded two hydrogen atoms to the one oxygen atom that was added. This is because we had checked the “Adjust Hydrogens” option. If we want to work with anions/cationswe should uncheck this box and then you can bond the added elements by left clicking the added atoms and dragging the cursor to another atom. Clicking on the created bond will change its type (double, triple etc.).
Press F10 to enter the Manipulation Tool mode. While in this mode, right click will allow you to pan the view, zoom in/out is performed using the mouse wheel, and view ordination can be adjusted by dragging the cursors in the drawing area. Double clicking the molecule will automatically center the view on it.
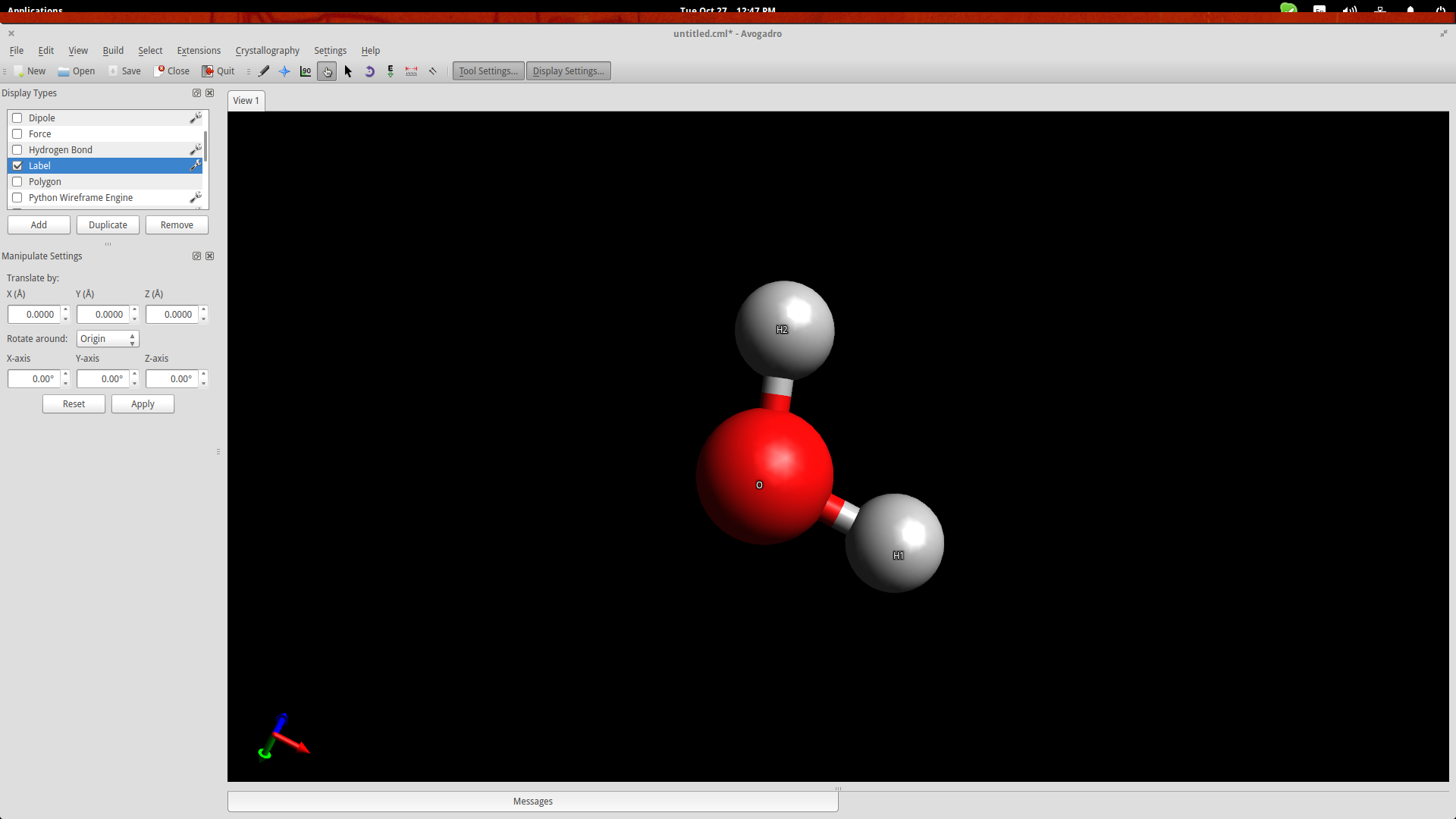
While in the same mode, you may also drag the individual atoms to change their position in space.
However, atoms form molecules in a particular way and all bonds have specific distances and energies. You can set those accurately by using the measurement tool. Invoke it by pressing F12 and then select the three atoms with the second being the oxygen atom because the second point acts as the reference point for the angle measurement.
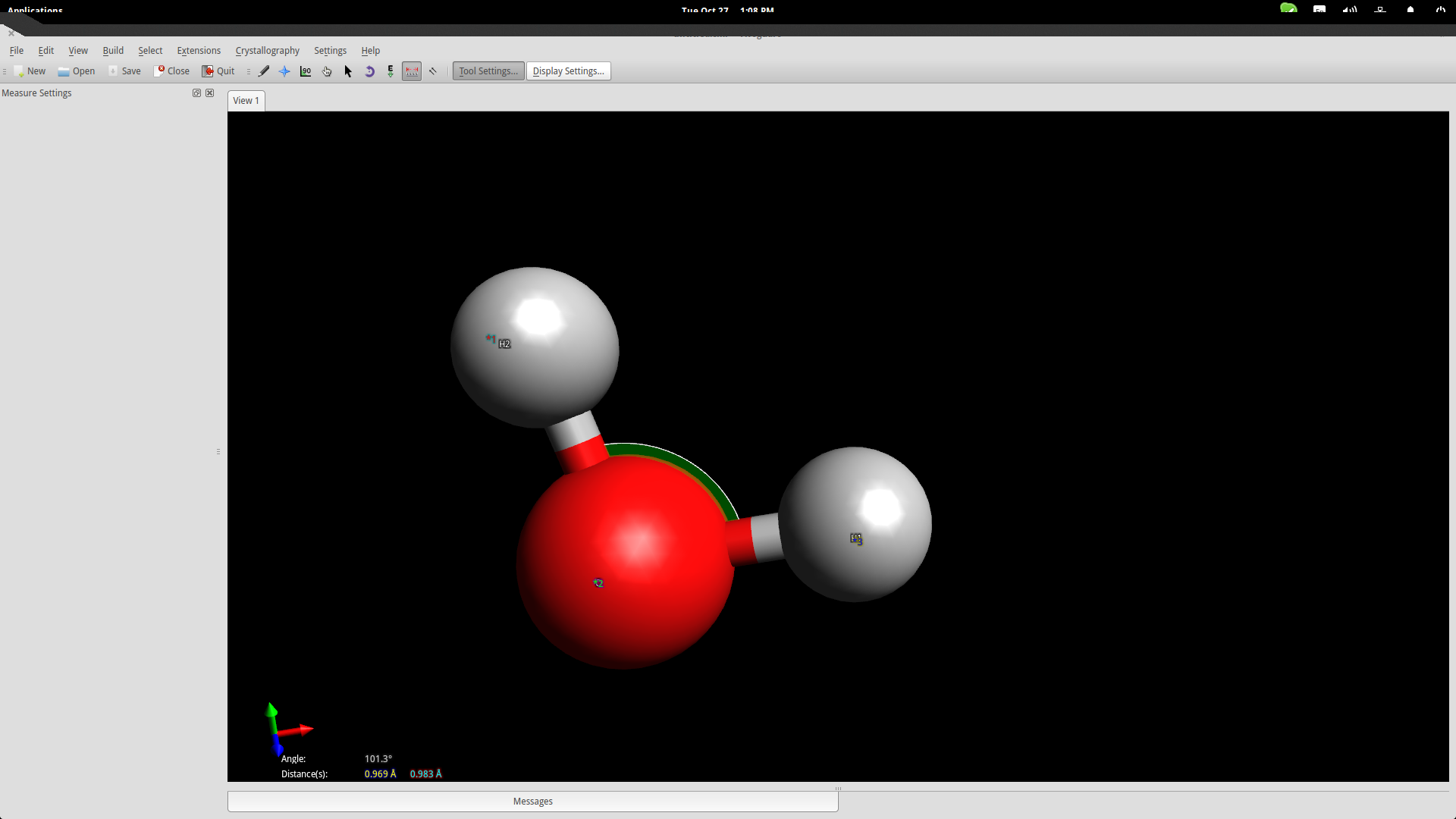
The bond distances seem to be a little bit longer than in reality, while the angle is slightly less than it should be, but we are within the accepted range for education purposes so let’s proceed to the next step.
Force Simulation
We added two water molecules in order to simulate the bond that is naturally formed between them thanks to their bipolar nature. Press the “Auto-Optimization Tool” button right below the top menu and then click the “Start” button on the left side of the interface to start the simulation. I used the “steepest descent” algorithm, which aligned the two molecules as shown in the screenshot below:
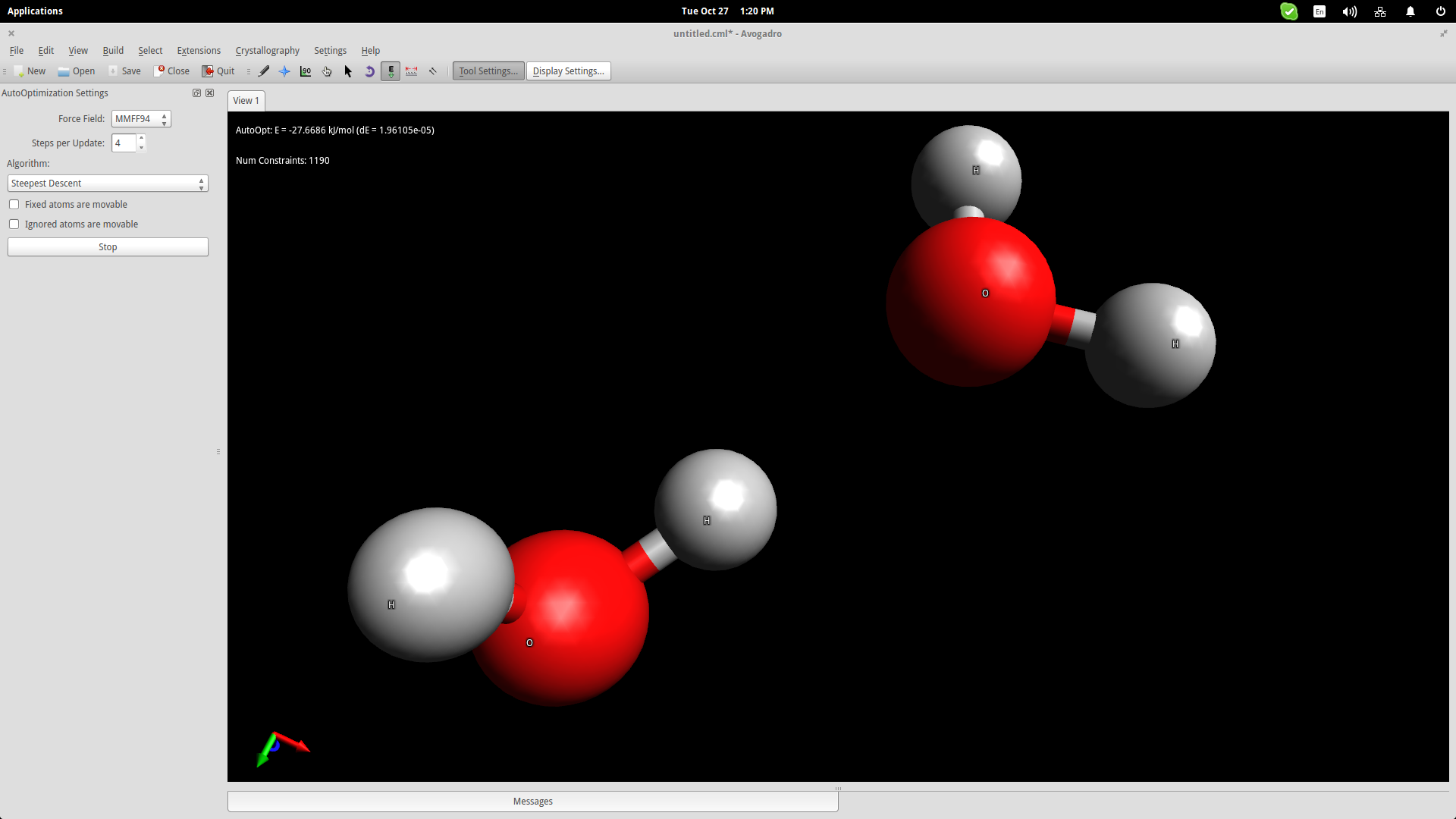
To show this better, we will use an electrostatic potential depiction on the two molecules. Go to the top panel and navigate on Extensions->Create Surfaces. On the window that will open up, choose the Electrostatic Potential option on the “Color By” menu and click on the “Calculate” button.
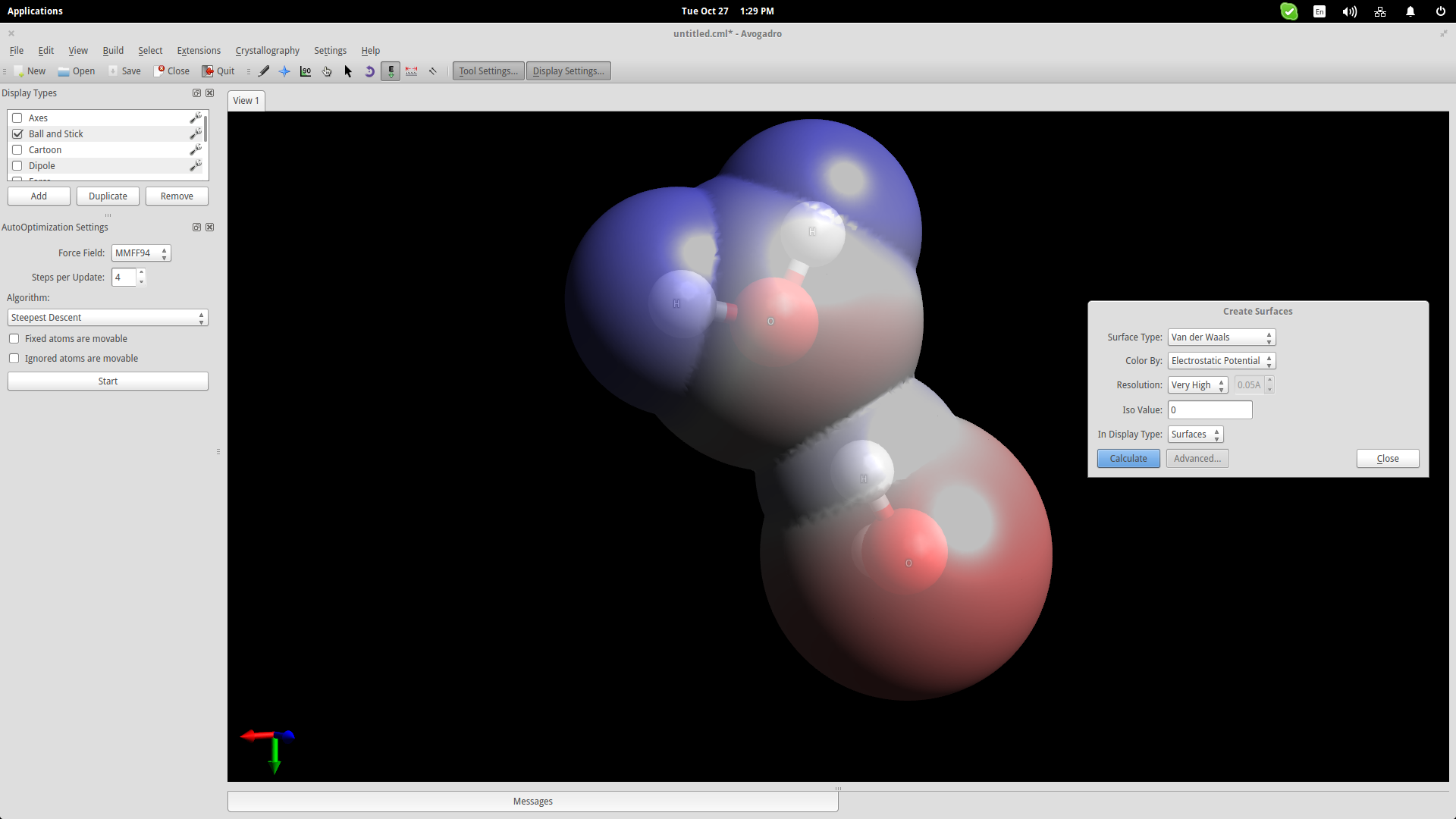
It is now clear that the hydrogen atom of one of the water molecules is bonding electro-statically with the oxygen atom of the second molecule. This helps students understand the nature of water molecules and why water is such a good dissolver.
DNA/RNA
Another useful educational feature of Avogadro (among the hundreds really) is the auto-generation of nucleic acids like DNA and RNA. Navigate to Build->Insert->DNA/RNA… on the top panel and add your own sequence and press the “Insert” button to insert the model in the drawing space.
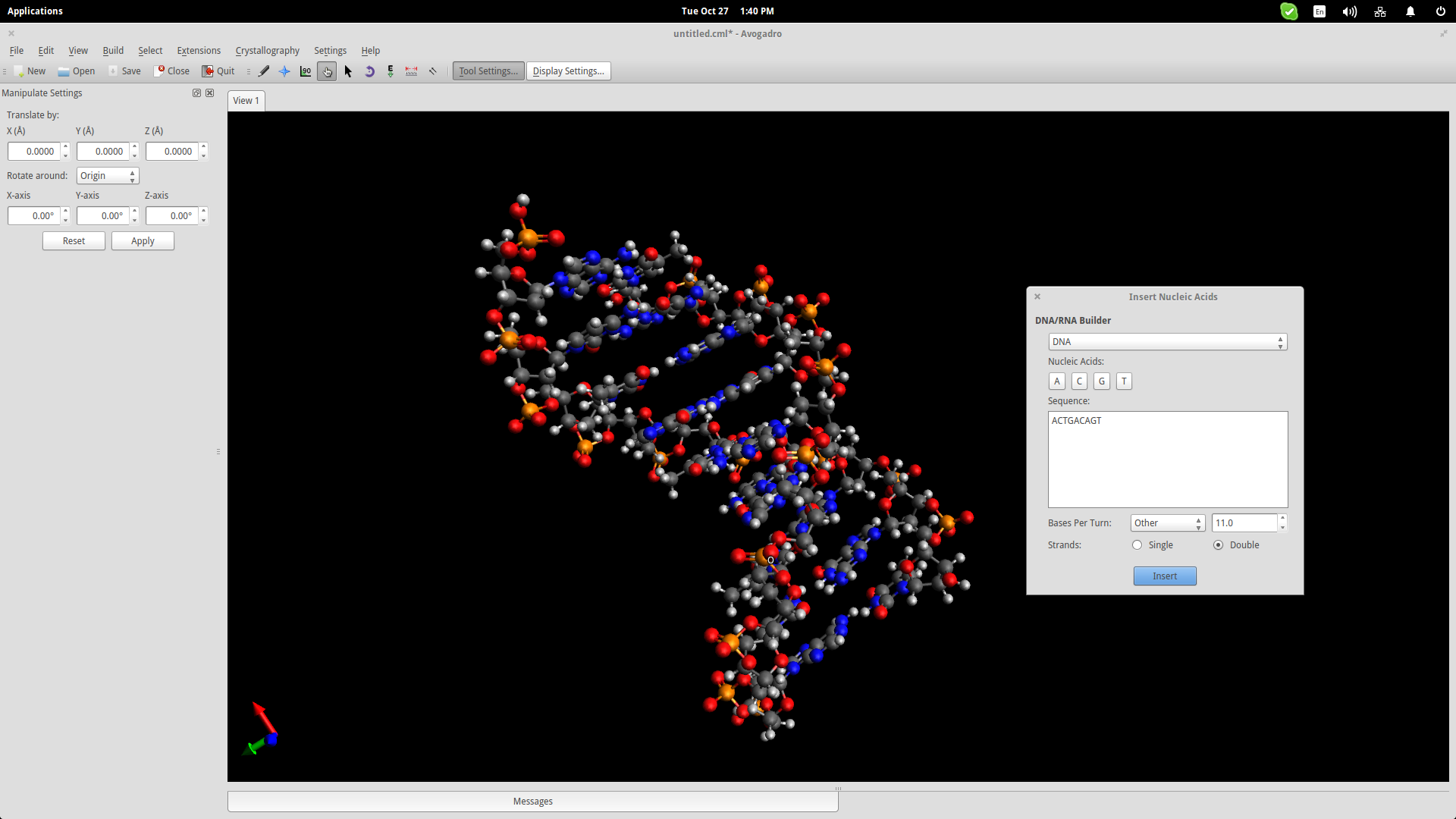
This could help students better understand the form of the genetic code of all living beings in the planet.
Crystal Generator/Viewer
One more thing that could be useful for educators is Avogadro’s crystal generator/viewer. Go to the top panel menu and click on Crystallography->Add Unit Cell. This will add a cube in the drawing area. On the top right, you may change the cube size parameters according to what you want to create. Add the base molecules in the cell and then go to Crystallography->Spacegroup->Fill Unit Cell and select the specified type from the list.
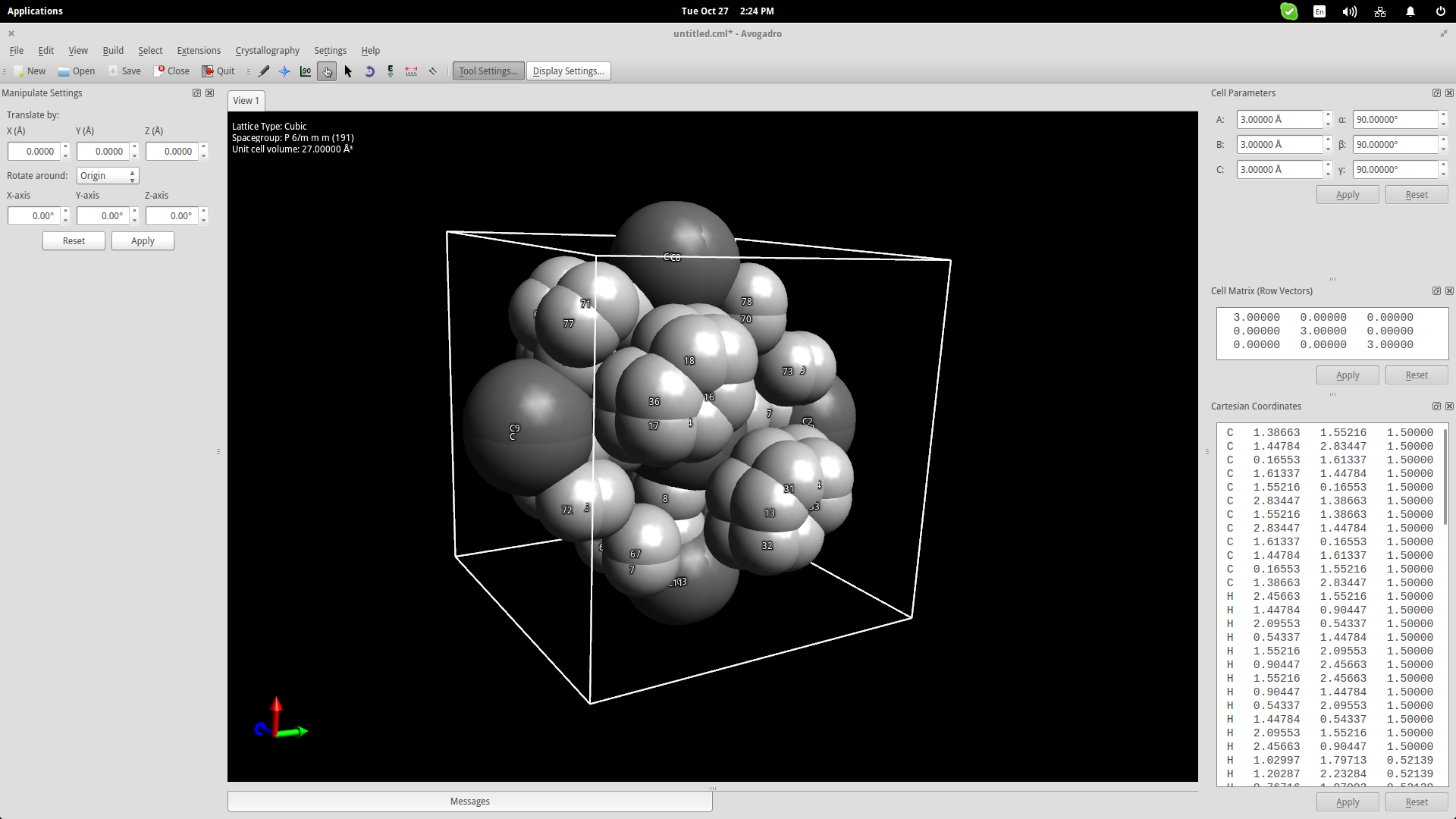
—
Avogadro is an advanced molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It offers flexible high quality rendering and a powerful plugin architecture.
Avogadro on cloud for AWS
Features
Avogadro Features include:
- Supports multi-threaded rendering and computation.
- “Auto-optimize” tool which allows you to continue to build and modify, during molecular mechanics optimization.
- Built-in molecular mechanics (including MMFF94 and UFF).
- Input generation for Gaussian and GAMESS-US, with more packages to come.
- Support for crystallographic unit cells.
- Visualization of isosurfaces and orbitals, including Gaussian cubes, OpenDX, and Gaussian fchk files.
- Open Babel import of files.
- Plugin architecture for developers, including rendering, interactive tools, commands, and Python scripts.
- Interfaces to many common computational packages.
- Embedded Python interpreter.
- Well defined public API, library and Python bindings for development.
- Translations into French, German, Italian, Russian, Spanish, Chinese and others.
Major Features of Avogadro
- Cross-Platform: Molecular builder/editor for Windows, Linux, and Mac OS X.
- Free, Open Source: Easy to install and all source code is available under the GNU GPL.
- International: Translations into Chinese, French, German, Italian, Russian, Spanish, and others, with more languages to come.
- Intuitive: Built to work easily for students and advanced researchers both.
- Fast: Supports multi-threaded rendering and computation.
- Extensible: Plugin architecture for developers, including rendering, interactive tools, commands, and Python scripts.
- Flexible: Features include Open Babel import of chemical files, input generation for multiple computational chemistry packages, crystallography, and biomolecules.
AWS
Installation Instructions For Windows
Note: How to find PublicDNS in AWS
Step 1) RDP Connection: To connect to the deployed instance, Please follow Instructions to Connect to Windows instance on AWS Cloud
1) Connect to the virtual machine using following RDP credentials:
- Hostname: PublicDNS / IP of machine
- Port : 3389
Username: To connect to the operating system, use RDP and the username is Administrator.
Password: Please Click here to know how to get password .
Step 2) Click the Windows “Start” button and select “All Programs” and then point to Avogadro .
Step 3) Other Information:
1.Default installation path: will be in your root folder “C:\Program Files (x86)\Avogadro”
2.Default ports:
- Windows Machines: RDP Port – 3389
- Http: 80
- Https: 443
Note: Click on Desktop icon – Press start then App will open in browser.
Configure custom inbound and outbound rules using this link
Installation Ste p by Step Screenshots

Videos
